
Drop-seq – Getting to the core of single cells one drop at a time
Without a doubt, in the last few years single cell research has developed into one of the most successful emerging fields in clinical and academic research. Since the establishment of single cell RNA-sequencing (scRNA-Seq) 10 years ago (1), a plethora of new single cell applications and technologies have made their way into our labs.
One of the most widely used techniques is Drop-seq, a microfluidics-based approach for scRNA-Seq (2). In the following, I will explain the ins and outs of Drop-seq as well as some of the most recent advances in this technology.
What is Drop-seq? An introduction
Already a Drop-seq pro? Feel free to skip to this chapter, for everyone else this will be valuable background for following this blog post.
Drop-seq is a method that was originally developed in 2015 by Macosko et al. In Drop-seq, single cells are encapsulated alongside beads in droplets (Figure 1). The beads have surfaces coated with oligonucleotides containing a (dT)30 stretch allowing for the capture of polyadenylated mRNA. After emulsion breakage, mRNA is subsequently reverse transcribed into cDNA. Each bead also carries a unique DNA barcode that identifies not only individual cells, but also individual mRNA transcripts derived from any given cell.
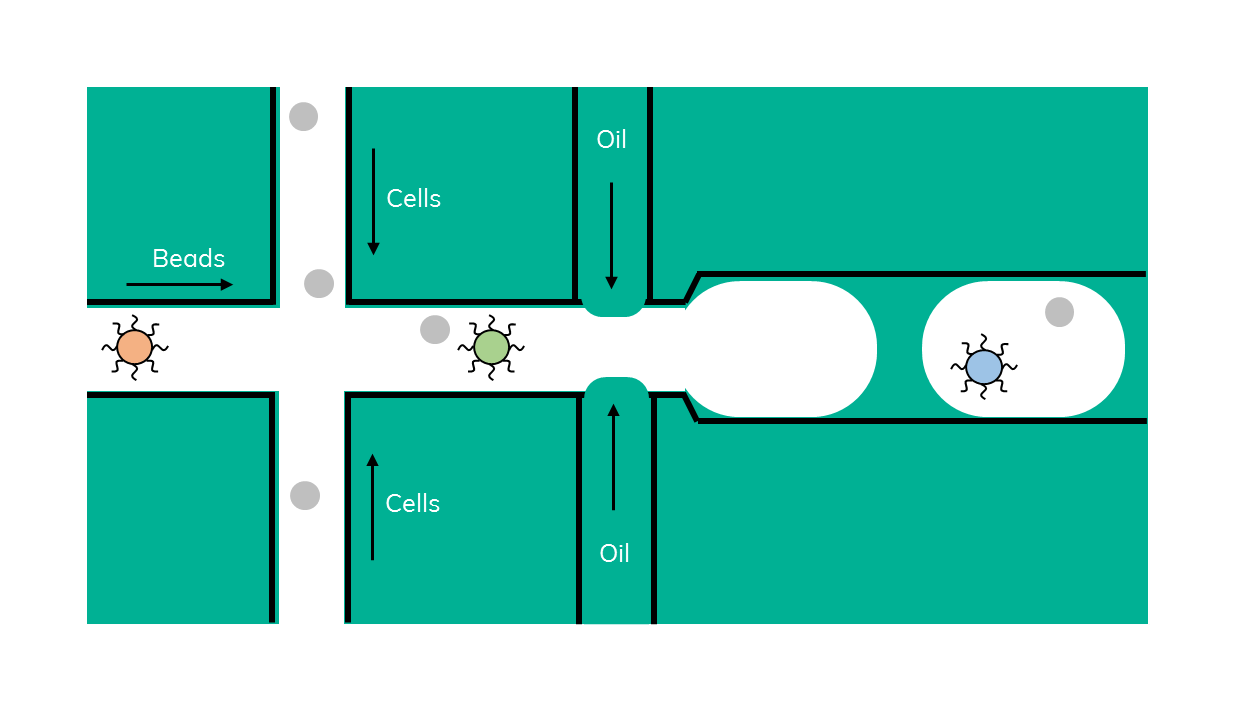
At around the same time, inDrop (Figure 2) was developed, a similar method that uses barcoded beads in lysis buffer that are encapsulated with a cell and a reverse transcription (RT) reaction mix (3). Inside the droplet, cells are being lysed, mRNA is bound by the bead and, in contrast to Drop-seq, immediately reverse transcribed inside the emulsion.
Drop-seq and you shall find – the advantages of using Drop-seq
Since its publication in 2015, Drop-seq has matured into an easy-to-use, high-throughput method that’s accessible for everyone. The method has a few unique advantages that give it a head start when it comes to single cell sequencing.
Small budget? – Drop-seq is your friend!
Let’s face it, single cell research is expensive. Accordingly, any methodology worth its salt must make generating single cell libraries as cost-effective as possible. Saving cost on reagents is one of the main ways to reduce overall spending. Drop-seq reagents, if sourced by the user, such as those available to you with the Nadia Platform, are actually quite affordable at about 6 cents per cell compared to the average cost of between 15 cents and $4 per cell for other methods on the market (4).
Another major cost factor for scRNA-Seq is the sequencing cost. The number of cells analysed per sample and the number of sequencing reads per cell will greatly affect NGS cost. Drop-seq allows users to pick any given number of cells between 100 and 6000 to be analysed from a sample by subsampling, allowing the user to only sequence the number of cells required. At the same time, required sequencing depth per cell are low compared to other methods (5).
A little more reaction please – why you should separate cell lysis from the reverse transcription
One of the main differences between Drop-seq and inDrop is the way in which RT is done. In inDrop, the RT is performed inside the droplet and therefore its reagents must be contained alongside the cell lysis solution and during the consecutive binding of the mRNA to the beads. In contrast, Drop-seq RT is performed as an additional step after mRNA-capture beads are released from the emulsion.
So, why would anyone perform RT outside of a droplet I hear you ask? If you try performing many reactions in the same reaction vessel (such as a droplet), conditions must be finely tuned to make all work. Even if this is achieved, it’s likely to be more of a compromise rather than an optimal condition for both RT and lysis. The lysis might not be as efficient, or the capture of mRNA is reduced. This is especially relevant when working with cells that are not bog-standard and require some tweaking of lysis conditions. When accounting for RT reagents that are sensitive to detergents used to lyse cells, lysis condition tweaking becomes borderline impossible (6). Another big advantage of separating lysis and RT is that it makes subsampling possible. A potent technique that enables further cost savings.
Taste before you waste – the benefits of subsampling your data with drop-seq
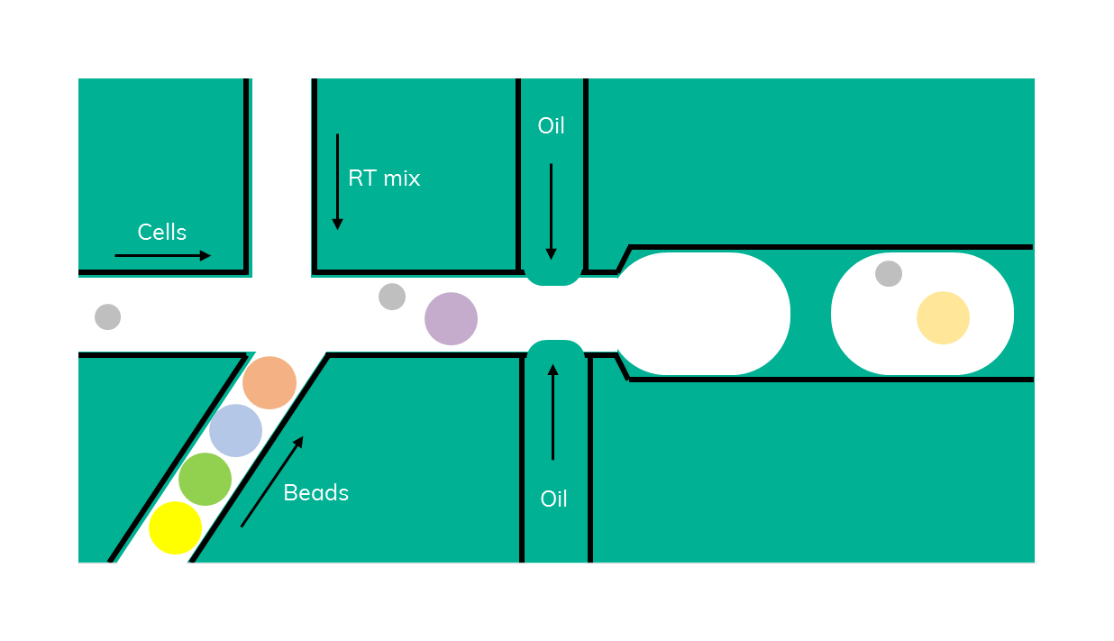
Subsampling, a term that those working in bioinformatics are well familiar with, is often used in the field of data analysis to compare available pipelines. Comparing pipelines using a full dataset would use up extensive computational resources. This is mirrored in single cell research. Just imagine, every time you capture a couple of thousand cells, all those cells would require sequencing. This would very quickly eat up hard earned grant money. By testing a smaller subset of cell transcriptomes first and, importantly, in their full complexity before committing to sequencing, the full sample is much more cost effective (Figure 3). This, however, only works if the amplification step (PCR or otherwise) is carried out on cDNA that is still bound to beads.
In Drop-seq, this is the case: following RT, which is done in bulk on all beads, PCR amplification starts from bead-bound cDNA. Users can choose the number of cells they wish to sequence by picking a certain number of beads to undergo PCR. This will enable them to retain information about complexity because amplification happens on the entirety of a single cell transcriptome.(Figure 3). This allows the user to test a representative subset of cells to gain insight into expression levels of the full sample.
In inDrop, the cDNA is released from the beads inside the droplets following RT. When the emulsion is broken to recover the cDNA, all the captured single-cell transcriptomes are mixed together, and amplification is carried out on mixed cDNA. Subsampling at this point would equate to taking a little bit of cDNA originating from each captured cell rather than full transcriptomes. This potentially biases the data towards genes with higher expression levels, as only a fraction of the total cDNA of each cell would be analysed (Figure 3).

What a difference a bead makes- the ability to use soft or hard beads
There are two main bead types used in scRNA-Seq, hard beads often made of polymethylmethacrylate and soft beads made from hydrogel (Figure 4).
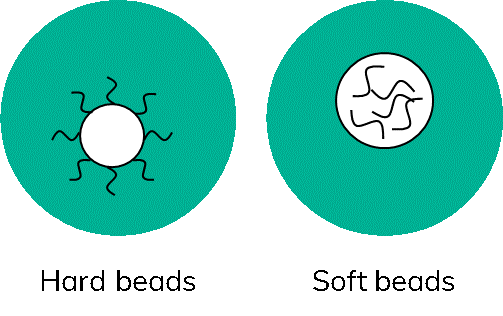
The name gives it away, hard beads are not deformable. Consequently, only so many beads can be passed through a microfluidic junction at a time without clogging the channels. Furthermore, it is not desired to have more than one bead per droplet as this could artificially inflate the numbers of cells detected. Therefore, working with hard beads will always require some form of limiting dilution.
Newer systems can obtain as many as 1 bead per 10 droplets, enabling the capture of about 10% of all cells loaded. Soft beads, on the other hand are made of a more flexible, gel-like material, that will squeeze through channels and junctions. This enables something called super-poisson loading; whereby the user could theoretically have 1 bead per droplet, with methods such as inDrop achieving an 80 to 90% cell capture rate (7). However, in reality, commercial systems typically achieve around 50% cell capture (8).
The main advantage of using hard beads is the price, as the cost per cell library is typically low and justifies the lower capture rate (9,10). In contrast, soft beads are useful specifically only if a limited number of cells are available, such as a couple of thousand cells. Descending further in terms of throughput, with decreasing amounts of cells, microfluidic approaches become less useful and well plate-based methods may be more suitable. Typically, however, researchers have thousands of cells available for analysis, meaning the capture rate is not the limiting factor. The real power of Drop-seq is that the method itself works independently of the bead type used. It is a highly flexible methodology that can use hard or soft beads according to user’s needs.
When every cell counts – flexibility with the Nadia Instrument
Most commercial systems take a one-size-fits-all approach and only leave minimal room for modification or optimization of the capture conditions beyond the cellular input (7). This is especially relevant when research questions require capturing samples with low cell input as well as optimizations such as altered lysis conditions.
Indeed, the solution might be close at hand. We have been working on a Drop-seq workflow using soft beads; combining both high cell capture rates with the flexibility of the scRNA-Seq on Nadia protocol (Figure 5).
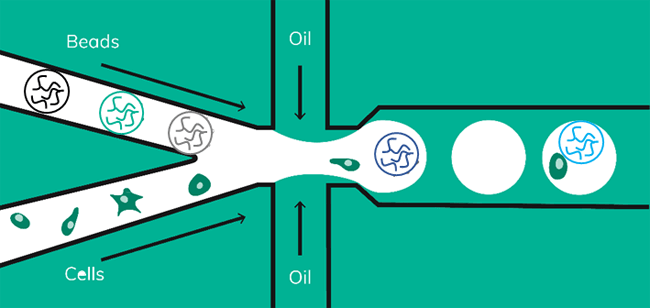
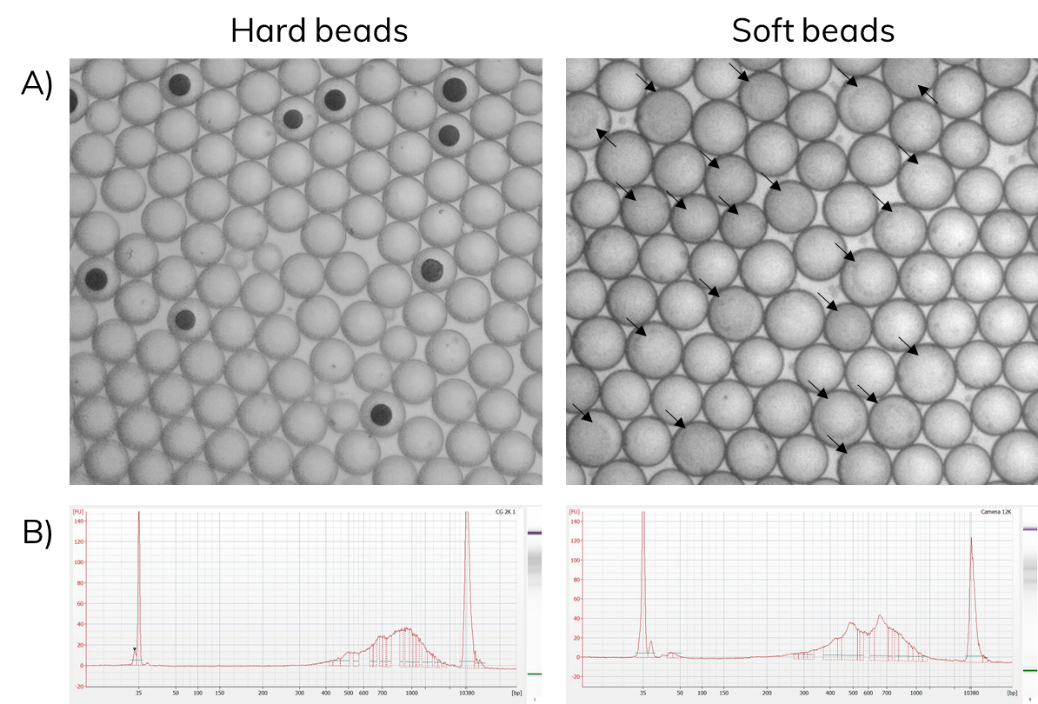
Barcoded soft beads were encapsulated alongside cells. Then, beads were recovered from the emulsion and libraries were amplified as described in the scRNA-Seq on Nadia protocol. The use of soft beads increased the cell capture rates from 10% to about 70% and produced libraries comparable to the standard Drop-seq protocol using hard beads from Chemgenes (Figure 6).
Summary
The Drop-seq method has received huge traction in the field given its high throughput coupled with low cost per cell. The method offers unique advantages such as high flexibility on lysis conditions and the option to subsample your cells. The main perceived drawback of the method, the low capture rate, is only due to the bead type used in the original published method. When combined with soft beads, Drop-seq delivers the ultimate combination of high cell capture rates and increased flexibility, opening scRNA-Seq up for everyone.
Find out how these advantages are available to you with scRNA-Seq on the Nadia Instrument

Further reading
Learn more about dropSeqPipe work flow in our blog post
Or read this how to guide:
References
- Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods. 2009 May;6(5):377–82.
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell. 2015 May 21;161(5):1202–14.
- Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell. 2015 May 21;161(5):1187–201.
- ABRF Single-Cell RNA-Seq Platform Comparison Finds Similar Performance, Drawbacks for Each [Internet]. GenomeWeb. [cited 2019 Sep 26]. Available from: https://www.genomeweb.com/sequencing/abrf-single-cell-rna-seq-platform-comparison-finds-similar-performance-drawbacks-each
- Zhang X, Li T, Liu F, Chen Y, Yao J, Li Z, et al. Comparative Analysis of Droplet-Based Ultra-High-Throughput Single-Cell RNA-Seq Systems. Mol Cell. 2019 03;73(1):130-142.e5.
- Hodne K, Weltzien F-A. Single-Cell Isolation and Gene Analysis: Pitfalls and Possibilities. Int J Mol Sci. 2015 Nov 10;16(11):26832–49.
- Salomon R, Kaczorowski D, Valdes-Mora F, Nordon RE, Neild A, Farbehi N, et al. Droplet-based single cell RNAseq tools: a practical guide. Lab Chip. 2019 May 14;19(10):1706–27.
- Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017 Jan 16;8:14049.
- AlJanahi AA, Danielsen M, Dunbar CE. An Introduction to the Analysis of Single-Cell RNA-Sequencing Data. Mol Ther – Methods Clin Dev. 2018 Sep 21;10:189–96.
- Ziegenhain C, Vieth B, Parekh S, Reinius B, Guillaumet-Adkins A, Smets M, et al. Comparative Analysis of Single-Cell RNA Sequencing Methods. Mol Cell. 2017 Feb 16;65(4):631-643.e4.